
Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
Поліміозит і дерматоміозит: причини, симптоми, діагностика, лікування
Медичний експерт статті

Останній перегляд: 05.07.2025
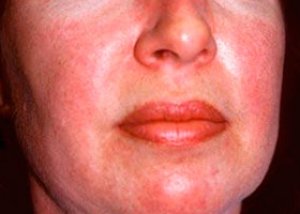
Поліміозит та дерматоміозит – це рідкісні системні ревматичні захворювання, що характеризуються запальними та дегенеративними змінами в м’язах (поліміозит) або м’язах і шкірі (дерматоміозит). Найбільш специфічним шкірним проявом є геліотропний висип.
Ураження м’язів симетричне та включає слабкість, певну болючість та подальшу атрофію м’язів проксимального відділу тазового пояса. Ускладнення можуть включати ураження внутрішніх органів та злоякісні новоутворення. Діагноз ґрунтується на клінічній картині та оцінці м’язової дисфункції шляхом вимірювання рівня ферментів, проведення МРТ, електроміографії та біопсії м’язів. Лікування включає глюкокортикоїди, іноді в поєднанні з імуносупресантами або внутрішньовенними імуноглобулінами.
Жінки хворіють вдвічі частіше за чоловіків. Захворювання може виникнути в будь-якому віці, але найчастіше виявляється в діапазоні від 40 до 60 років; у дітей – від 5 до 15 років.
 [
[ Що викликає дерматоміозит та поліміозит?
Вважається, що причиною захворювання є аутоімунна реакція на м'язову тканину у генетично схильних осіб. Захворювання частіше зустрічається за наявності обтяженого сімейного анамнезу та носіїв певних HLA-антигенів (DR3, DR52, DR56). Можливими тригерами є вірусний міозит та злоякісні новоутворення. Є повідомлення про виявлення структур, подібних до пікорнавірусів, у м'язових клітинах; крім того, віруси можуть викликати подібні захворювання у тварин. Зв'язок злоякісних пухлин з дерматоміозитом (набагато рідше, ніж з поліміозитом) свідчить про те, що ріст пухлини також може бути тригером розвитку захворювання в результаті ініціації аутоімунних реакцій на спільні антигени пухлини та м'язової тканини.
Відкладення IgM, IgG та третього компонента комплементу виявляються в стінках кровоносних судин скелетних м'язів; це особливо характерно для дерматоміозиту у дітей. У пацієнтів з поліміозитом також можуть розвиватися інші аутоімунні процеси.
Патофізіологія дерматоміозиту та поліміозиту
Патологічні зміни включають пошкодження та атрофію клітин на тлі запалення різного ступеня тяжкості. М'язи верхніх і нижніх кінцівок, а також обличчя, уражаються меншою мірою, ніж інші скелетні м'язи. Пошкодження вісцеральних м'язів глотки та верхніх відділів стравоходу, рідше серця, шлунка або кишечника, може призвести до порушення функції вищезгаданих органів. Висока концентрація міоглобіну внаслідок рабдоміолізу може спричинити ураження нирок. Також можуть виникати запальні зміни в суглобах і легенях, особливо у пацієнтів, які мають антисинтетазні антитіла.
Симптоми дерматоміозиту та поліміозиту
Початок поліміозиту може бути гострим (особливо у дітей) або підгострим (частіше у дорослих). Гостра вірусна інфекція іноді передує або є пусковим механізмом для прояву захворювання, найпоширенішими проявами якого є слабкість проксимальних м'язів або шкірні висипання. Біль виражений меншою мірою, ніж слабкість. Можуть розвинутися поліартралгія, феномен Рейно, дисфагія, порушення функції легень, загальні симптоми (підвищення температури тіла, зниження маси тіла, слабкість). Феномен Рейно часто зустрічається у пацієнтів із супутніми захворюваннями сполучної тканини.
М'язова слабкість може прогресувати протягом кількох тижнів або місяців. Однак для клінічного прояву м'язової слабкості має бути уражено щонайменше 50% м'язових волокон (таким чином, наявність м'язової слабкості вказує на прогресування міозиту). Пацієнти можуть відчувати труднощі з підняттям рук вище рівня плечей, ходьбою по сходах або вставання з положення сидячи. Через виражену слабкість м'язів тазу та плечового пояса пацієнти можуть бути прикуті до інвалідного візка або ліжка. Якщо уражені згиначі шиї, підняти голову з подушки стає неможливим. Ураження м'язів глотки та верхньої частини стравоходу призводить до порушень ковтання та відрижки. М'язи нижніх, верхніх кінцівок та обличчя зазвичай не уражаються. Однак можуть розвиватися контрактури кінцівок.
Шкірні висипання, що спостерігаються при дерматоміозиті, зазвичай темного кольору та еритематозні. Характерним також є періорбітальний набряк фіолетового кольору (геліотропний висип). Шкірні висипання можуть бути дещо піднятими над рівнем шкіри та бути гладкими або лускатими; локалізація висипань - лоб, шия, плечі, груди, спина, передпліччя, нижні частини гомілок, брови, колінні ділянки, медіальні кісточки, тильні поверхні міжфалангових та п'ястнофалангових суглобів, з латерального боку (симптом Готтрона). Можлива гіперемія основи або периферії нігтів. На шкірі латеральної поверхні пальців може розвинутися десквамативний дерматит, що супроводжується тріщинами. Первинні ураження шкіри часто проходять без наслідків, але можуть призвести до вторинних змін, таких як темна пігментація, атрофія, рубцювання або вітиліго. Можуть розвиватися підшкірні кальцифікати, особливо у дітей.
Приблизно у 30% пацієнтів розвивається поліартралгія або поліартрит, часто супроводжувані набряком та випотом у суглобах. Однак тяжкість суглобових проявів невелика. Вони частіше виникають, коли у пацієнтів виявляються антитіла до Jo-1 або інших синтетаз.
Ураження внутрішніх органів (крім глотки та верхньої частини стравоходу) трапляється при поліміозиті рідше, ніж при інших ревматичних захворюваннях (зокрема, при системному вовчазному черевному вовчазі та системній склеродермії). Рідко, особливо при антисинтетазному синдромі, захворювання проявляється як інтерстиціальний пневмоніт (у вигляді задишки та кашлю). Можуть розвиватися серцеві аритмії та порушення провідності, але вони зазвичай протікають безсимптомно. Шлунково-кишкові прояви частіше зустрічаються у дітей, які також хворіють на васкуліт, і можуть включати блювання з кров’ю, мелену та перфорацію кишечника.
Де болить?
Класифікація поліміозиту
Існує 5 типів поліміозиту.
- Первинний ідіопатичний поліміозит, який може виникнути в будь-якому віці, не вражає шкіру.
- Первинний ідіопатичний дерматоміозит схожий на первинний ідіопатичний поліміозит, але вражає шкіру.
- Поліміозит та дерматоміозит, пов'язані зі злоякісними новоутвореннями, можуть виникати у пацієнтів будь-якого віку; їх розвиток найчастіше спостерігається у пацієнтів літнього віку, а також у пацієнтів з іншими захворюваннями сполучної тканини. Розвиток злоякісних новоутворень може спостерігатися як протягом 2 років до, так і протягом 2 років після початку міозиту.
- Дитячий поліміозит або дерматоміозит пов'язаний із системним васкулітом.
- Поліміозит та дерматоміозит також можуть виникати у пацієнтів з іншими захворюваннями сполучної тканини, найчастіше прогресуючим системним склерозом, змішаним захворюванням сполучної тканини та системним вченим черевним вовчаком.
Включення міозиту м'язів тулуба до групи поліміозитів є неправильним, оскільки останній є окремим захворюванням, що характеризується клінічними проявами, подібними до проявів хронічного ідіопатичного поліміозиту. Однак він розвивається у літньому віці, часто вражає м'язи дистальних частин тіла (наприклад, верхніх і нижніх кінцівок), має триваліший перебіг, гірше реагує на лікування та характеризується типовою гістологічною картиною.
Діагностика дерматоміозиту та поліміозиту
Поліміозит слід підозрювати у пацієнтів зі скаргами на слабкість проксимальних м'язів, з болючістю або без неї. Обстеження на дерматоміозит необхідне у пацієнтів зі скаргами на висип, що нагадує геліотроп або симптом Готтрона, а також у пацієнтів з проявами поліміозиту в поєднанні з будь-якими ураженнями шкіри, що відповідають дерматоміозиту. Клінічні прояви поліміозиту та дерматоміозиту можуть нагадувати прояви системного склерозу або, рідше, ВКВ чи васкуліту. Впевненість у діагнозі підвищується, якщо відповідати якомога більшій кількості з наступних п'яти критеріїв:
- слабкість проксимальних м'язів;
- характерні висипання на шкірі;
- підвищена активність ферментів м’язової тканини (креатинкінази або, за відсутності підвищення її активності, амінотрансфераз чи альдолази);
- характерні зміни на міографії або МРТ;
- характерні гістологічні зміни в біопсії м'язової тканини (абсолютний критерій).
Біопсія м'язів може виключити деякі клінічно подібні стани, такі як міозит м'язів тулуба та рабдоміоліз, спричинений вірусною інфекцією. Зміни, виявлені гістологічним дослідженням, можуть відрізнятися, але типовими є хронічне запалення, вогнища дегенерації та регенерації м'язів. Перед початком потенційно токсичного лікування необхідна точна діагностика (зазвичай шляхом гістологічної верифікації). МРТ може виявити вогнища набряку та запалення в м'язах, а потім провести їх цілеспрямовану біопсію.
Лабораторні дослідження можуть підтвердити або, навпаки, виключити підозру щодо наявності захворювання, а також корисні для оцінки його тяжкості, можливості поєднання з іншою подібною патологією та діагностики ускладнень. Незважаючи на те, що антинуклеарні антитіла виявляються у деяких пацієнтів, це явище більш типове для інших захворювань сполучної тканини. Близько 60% пацієнтів мають антитіла до антигену ядер (PM-1) або цілих клітин тимуса та Jo-1. Роль аутоантитіл у патогенезі захворювання залишається неясною, хоча відомо, що антитіла до Jo-1 є специфічним маркером антисинтетазного синдрому, включаючи фіброзуючий альвеоліт, легеневий фіброз, артрит та феномен Рейно.
Періодична оцінка активності креатинкінази корисна для моніторингу лікування. Однак у пацієнтів з тяжкою м'язовою атрофією активність ферменту може бути нормальною, незважаючи на наявність хронічного активного міозиту. МРТ, біопсія м'язів або підвищена активність креатинкінази часто допомагають диференціювати рецидив поліміозиту та міопатію, індуковану глюкокортикоїдами.
Оскільки у багатьох пацієнтів є недіагностовані злоякісні новоутворення, деякі автори рекомендують проводити скринінг усіх дорослих з дерматоміозитом та тих, хто має поліміозит, віком понад 60 років, за таким графіком: фізикальне обстеження, включаючи обстеження молочних залоз, органів малого тазу та прямої кишки (включаючи аналіз калу на приховану кров); загальний аналіз крові; біохімічний аналіз крові; мамографія; визначення карциноембріонального антигену; аналіз сечі; рентгенографія грудної клітки. Деякі автори ставлять під сумнів необхідність такого скринінгу у молодших пацієнтів, які не мають клінічних ознак злоякісного новоутворення.
Що потрібно обстежити?
Як обстежувати?
Лікування дерматоміозиту та поліміозиту
Доки запалення не зникне, фізичну активність слід обмежувати. Глюкокортикоїди є препаратами першої лінії. У гострій стадії захворювання дорослим пацієнтам слід призначати преднізолон (перорально) у дозі від 40 до 60 мг на добу. Регулярне визначення активності креатинкінази є раннім показником ефективності: у більшості пацієнтів зниження або нормалізація відзначається протягом 6-12 тижнів після збільшення м'язової сили. Після нормалізації активності ферментів дозу преднізолону зменшують: спочатку приблизно на 2,5 мг на добу протягом тижня, потім швидше; якщо підвищена активність м'язових ферментів рецидивує, дозу гормону знову збільшують. Одужані пацієнти можуть обходитися без глюкокортикоїдів, але найчастіше дорослим пацієнтам потрібна тривала глюкокортикоїдна терапія (10-15 мг преднізолону на добу). Початкова доза преднізолону для дітей становить 30-60 мг/м2 один раз на добу. За наявності ремісії > 1 року у дітей терапію глюкокортикоїдами можна припинити.
У деяких випадках у пацієнтів, які отримують високі дози глюкокортикоїдів, спостерігається раптове посилення м’язової слабкості, що може бути пов’язано з розвитком глюкокортикоїдної міопатії.
У разі неадекватної відповіді на лікування глюкокортикоїдами, а також у разі розвитку глюкокортикоїдної міопатії або інших ускладнень, що потребують зниження дози або припинення прийому преднізолону, слід застосовувати імуносупресанти (метотрексат, циклофосфамід, азатіоприн, циклоспорин). Деякі пацієнти можуть отримувати лише метотрексат (зазвичай у дозах, що перевищують ті, що використовуються для лікування РА) більше 5 років. Внутрішньовенні імуноглобуліни можуть бути ефективними у пацієнтів, резистентних до медикаментозної терапії, але їх застосування збільшує вартість лікування.
Міозит, пов'язаний з первинними та метастатичними пухлинами, а також міозит м'язів тулуба, зазвичай менш стійкі до глюкокортикоїдної терапії. Ремісія міозиту, пов'язаного зі злоякісними пухлинами, можлива після видалення пухлини.
Який прогноз при дерматоміозиті та поліміозиті?
Тривала ремісія (і навіть клінічне одужання) понад 5 років спостерігається у більш ніж половини пацієнтів, які пройшли лікування; у дітей цей показник вищий. Рецидив, однак, може статися будь-коли. Загальна п'ятирічна виживаність становить 75%, у дітей вона вища. Причинами смерті у дорослих є тяжка та прогресуюча м'язова слабкість, дисфагія, порушення харчування, аспіраційна пневмонія або дихальна недостатність внаслідок легеневих інфекцій. Поліміозит протікає важче та стійкіше до лікування, якщо є ураження серця та легень. Смерть у дітей може настати внаслідок кишкового васкуліту. Загальний прогноз захворювання також визначається наявністю злоякісних новоутворень.