
Весь контент iLive перевіряється медичними експертами, щоб забезпечити максимально можливу точність і відповідність фактам.
У нас є строгі правила щодо вибору джерел інформації та ми посилаємося тільки на авторитетні сайти, академічні дослідницькі інститути і, по можливості, доведені медичні дослідження. Зверніть увагу, що цифри в дужках ([1], [2] і т. д.) є інтерактивними посиланнями на такі дослідження.
Якщо ви вважаєте, що який-небудь з наших матеріалів є неточним, застарілим або іншим чином сумнівним, виберіть його і натисніть Ctrl + Enter.
Лісенцефалія головного мозку
Медичний експерт статті

Останній перегляд: 12.07.2025
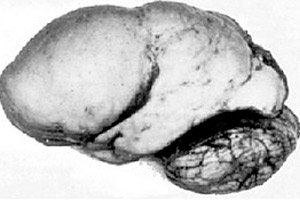
Серед органічних церебральних патологій виділяється така вроджена аномалія розвитку мозку, як лісенцефалія, суть якої полягає в майже гладкій поверхні кори його півкуль – з недостатньою кількістю звивин і борозен. [ 1 ]
При повній відсутності звивин визначається агірія, а наявність кількох широких плоских звивин називається пахігірією. Ці дефекти, як і деякі інші редукційні деформації мозку, мають код Q04.3 у МКХ-10.
Епідеміологія
Згідно зі статистикою рідкісних захворювань, на 100 тисяч новонароджених припадає 1-1,2 випадки лісенцефалії. [ 2 ], [ 3 ]
За деякими даними, у дітей із синдромом Міллера-Дікера спостерігається до 25-30% випадків класичної лісенцефалії; точкові мутації та делеції генів LIS1 та DCX виявляються майже у 85% пацієнтів. [ 4 ]
Генетичні дослідження 17 генів, пов'язаних з лісенцефалією, показали, що мутація або делеція LIS1 є причиною 40% пацієнтів, а 23% пов'язані з мутацією DCX, далі йдуть TUBA1A (5%) та DYNC1H1 (3%).[ 5 ]
Причини ліссенцефалії
Усі відомі причини формування кори головного мозку (cortex cerebri) майже або повністю без звивин і борозен, що збільшують «робочу площу» людського мозку та забезпечують «продуктивність» центральної нервової системи, пов’язані з порушеннями її перинатального розвитку. Тобто, у плода розвивається лісенцефалія. [ 6 ]
Нездатність формувати шари кори головного мозку плода при лісенцефалії є результатом аномальної міграції нейронів, що її утворюють, або передчасного припинення цього процесу.
Цей процес, який є важливим для цереброкортикального гістогенезу, відбувається в кілька етапів з 7-го по 18-й тиждень вагітності. А враховуючи його підвищену чутливість до генетичних мутацій, а також різних негативних фізичних, хімічних та біологічних впливів, будь-яке відхилення від норми може призвести до неправильної локалізації нейронів з можливим утворенням потовщеного шару сірої речовини кори без характерної структури. [ 7 ]
У деяких випадках лісенцефалія у дітей пов'язана з синдромами Міллера-Дікера, Вокера-Варбурга або Нормана-Робертса.
Читайте також – Вади розвитку мозку
Фактори ризику
Окрім мутацій у деяких генах, до факторів ризику народження дитини з таким важким дефектом належать кисневе голодування (гіпоксія) плода; недостатнє кровопостачання мозку (гіпоперфузія); гостре порушення мозкового кровообігу у вигляді перинатального інсульту; патології плаценти; вірусні інфекції вагітної (включаючи TORCH); [ 8 ] проблеми із загальним обміном речовин та функцією щитовидної залози; куріння, вживання алкоголю, психотропних та наркотичних речовин; вживання низки ліків; підвищений рівень радіації. [ 9 ]
Патогенез
Не всі випадки лісенцефалії мають патогенез, спричинений хромосомними аномаліями та генними мутаціями. Але відомі деякі гени, що кодують білки, що відіграють важливу роль у правильному русі нейробластів і нейронів вздовж клітин радіальної глії – для формування кори головного мозку. І мутації цих генів призводять до цієї патології. [ 10 ]
Зокрема, це спорадичні мутації (без спадковості) гена LIS1 на 17-й хромосомі, який регулює цитоплазматичний моторний білок мікротрубочок динеїн, а також гена DCX на Х-хромосомі, який кодує білок даблкортин (лісенцефалін-X). [ 11 ]. У першому випадку фахівці визначають класичну лісенцефалію (тип I), у другому – Х-зчеплену. [ 12 ]
Коли ген FLN1, що кодує фосфопротеїн філамін 1, видаляється, процес спрямованої міграції нейронів може взагалі не розпочатися, що призводить до повної відсутності звивин (агірії). [ 13 ]
Виявлено мутації в гені CDK5, який кодує фермент кіназу – каталізатор внутрішньоклітинного метаболізму, що регулює клітинний цикл у нейронах ЦНС та забезпечує їх нормальну міграцію під час пренатального формування структур мозку.
Аномальні зміни в гені RELN на 7-й хромосомі, що спричиняють дефекти кортикальної звивини при синдромі Нормана-Робертса, призводять до дефіциту позаклітинного глікопротеїну реліну, який необхідний для регуляції міграції та позиціонування нервових стовбурових клітин під час розвитку кори головного мозку.[ 14 ], [ 15 ], [ 16 ]
Ген ARX кодує неаристаленовий гомеобоксний білок, транскрипційний фактор, який відіграє важливу роль у передньому мозку та інших тканинах.[ 17 ] Діти з мутацією ARX мають інші симптоми, такі як відсутність частин мозку (агенезія мозолистого тіла), аномальні геніталії та тяжка епілепсія.[ 18 ],[ 19 ]
Кілька генів пов'язані з лісенцефалією. Ці гени включають VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A та CDK5.[ 20 ]
Цитомегаловірус (ЦМВ) пов'язують з розвитком лісенцефалії через зниження кровопостачання мозку плода. Тяжкість ЦМВ-інфекції залежить від гестаційного віку. Рання інфекція частіше спричиняє лісенцефалію, оскільки міграція нейронів відбувається на ранніх термінах вагітності.[ 21 ]
Крім того, механізм виникнення цієї аномалії включає неповне або пізніше припинення міграції нейронів з перивентрикулярної генеративної зони до кори головного мозку. І в таких випадках розвивається або неповна лісенцефалія, або пахігірія, при якій утворюється кілька широких борозен і звивин (але більшість з них відсутні).
Симптоми ліссенцефалії
Перші ознаки цієї патології (за відсутності раніше згаданих синдромів) можуть проявитися не одразу після народження, а через півтора-два місяці. І найчастіше спостерігаються такі клінічні симптоми лісенцефалії:
- м'язова гіпотонія, часто поєднується зі спастичним паралічем;
- судоми та генералізовані тоніко-клонічні судоми (у формі опістотонуса);
- важка розумова відсталість та затримка росту;
- порушення неврологічних та рухових функцій.
Проблеми з ковтанням спричиняють труднощі з годуванням дитини. [ 22 ]
Високий ступінь нейромоторних порушень часто проявляється тетраплегією – паралічем усіх кінцівок. Можлива деформація кистей, пальців рук або ніг.
При синдромі Нормана-Робертса з лісенцефалією I типу спостерігаються краніофаціальні аномалії: виражена мікроцефалія, низький нахил чола та виступаюча широка перенісся, широко розставлені очі (гіпертерлоризм), недорозвинення щелеп (мікрогнатія). [ 23 ]
Синдром Міллера-Дікера також може характеризуватися аномально малим розміром голови з широким, високим чолом і коротким носом, заглибленнями в скронях (бітемпоральними заглибленнями) та низько посадженими, деформованими вухами.
Синдром тяжкої лісенцефалії характеризується мікроцефалією, зменшенням розмірів очних яблук (мікрофтальмією), що поєднується з дисплазією сітківки, обструктивною гідроцефалією та відсутнім або гіпопластичним мозолистим тілом.
Ускладнення і наслідки
Серед ускладнень цієї аномалії фахівці називають порушення функції ковтання (дисфагію) та гастроезофагеальний рефлюкс; рефрактерну (неконтрольовану) епілепсію; часті інфекції верхніх дихальних шляхів; пневмонію (включаючи хронічну аспіраційну).
Немовлята з лісенцефалією можуть мати вроджені органічні проблеми з серцем у вигляді дефекту міжпередсердної перегородки або складного пороку серця з ціанозом (тетралогія Фалло). [ 24 ]
Наслідки постнатальної затримки розвитку в більшості випадків призводять до смерті протягом 24 місяців після народження.
Діагностика ліссенцефалії
Діагностика починається з фізичного огляду дитини, вивчення історії хвороби батьків та історії вагітності та пологів.
Під час вагітності може знадобитися безклітинне тестування фетальної ДНК, амніоцентез або взяття біопсії ворсин хоріона. [ 25 ] Для отримання додаткової інформації див. – Пренатальна діагностика вроджених захворювань
Інструментальна діагностика використовується для візуалізації структур мозку та оцінки їх функцій:
- комп'ютерна томографія головного мозку;
- магнітно-резонансна томографія (МРТ) головного мозку;
- електроенцефалограма (ЕЕГ). [ 26 ]
Під час вагітності лісенцефалію на УЗД плода після 20-21 тижня можна запідозрити при відсутності тім'яно-потиличної та п'яткової борозен та аномалії сильвієвої щілини головного мозку.
Диференціальна діагностика
Проводиться диференціальна діагностика з іншими синдромами вроджених вад головного мозку.
Існує понад 20 типів лісенцефалії, більшість з яких поділяються на 2 основні категорії: класична лісенцефалія (тип 1) та лісенцефалія типу «бруківка» (тип 2). Кожна категорія має подібні клінічні прояви, але різні генетичні мутації.[ 27 ]
Дослідження мозку при лісенцефалії I типу показує кору головного мозку з чотирма шарами замість шести, як у здорових пацієнтів, тоді як при лісенцефалії 2 типу кора головного мозку дезорганізована та виглядає горбистою або вузлуватою через повне зміщення кори головного мозку кластерами кортикальних нейронів, розділених гліомезенхімальною тканиною. У пацієнтів також спостерігалися м'язові та очні аномалії.
- Класична лісенцефалія (тип 1):
- LIS1: Ізольована лісенцефалія та синдром Міллера-Дікера (лісенцефалія, пов'язана з дисморфізмом обличчя). [ 28 ]
- LISX1: мутація гена DCX. Порівняно з лісенцефалією, спричиненою мутаціями LIS1, DCX демонструє шестишарову кору замість чотирьох.
- Ізольована лісенцефалія без інших відомих генетичних дефектів
- Лісенцефалія з бруківки (тип 2):
- Синдром Вокера-Варбурга
- Синдром Фукуями
- Захворювання м'язів, очей та мозку
- Інші типи не можна віднести до жодної з двох вищезгаданих груп:
- LIS2: синдром Нормана-Робертса, подібний до лісенцефалії I типу або синдрому Міллера-Дікера, але без делеції 17-ї хромосоми.
- LIS3
- LISX2
Мікролісенцефалія: це поєднання відсутності нормальної кіркової складки та аномально маленької голови. Діти з правильною лісенцефалією мають нормальний розмір голови при народженні. Дітям з маленьким розміром голови при народженні зазвичай ставлять діагноз мікролісенцефалія.
Також важливо розрізняти лісенцефалію та полімікрогірію, які є різними дефектами розвитку мозку.
До кого звернутись?
Лікування ліссенцефалії
Лісенцефалія — це невиліковний органічний дефект, тому можливе лише підтримуюче та симптоматичне лікування. [ 29 ]
Перш за все, це застосування протисудомних та протиепілептичних препаратів, а також встановлення гастростомічної трубки в шлунок (якщо дитина не може самостійно ковтати). Корисний масаж.
У випадках тяжкої гідроцефалії видаляють спинномозкову рідину.
Профілактика
Фахівці рекомендують майбутнім батькам звертатися за генетичною консультацією, а вагітним жінкам своєчасно ставати на облік до акушерів-гінекологів та проходити всі планові обстеження.
Прогноз
Для дітей з лісенцефалією прогноз залежить від її ступеня, але найчастіше розумовий розвиток дитини не перевищує рівня чотирьох-п'яти місяців. І всі діти з цим діагнозом страждають від важких психомоторних порушень та важколікувальної епілепсії. [ 30 ]
За даними NINDS (Американського національного інституту неврологічних розладів та інсульту), максимальна тривалість життя людей з лісенцефалією становить близько 10 років.